Пріонні захворювання — це група нейродегенеративних хвороб людини та тварин, до яких відносяться хвороба Крейтцфельдта—Якоба (ХКЯ), хвороба Герстманна—Штраусслера—Шейнкера (ХГШШ) у людей, та губчаста енцефалопатія великої рогатої худоби (ГЕВРХ, або «коров'ячий сказ») та скрепі у овець. Всі ці захворювання мають тривалий інкубаційний період, але зазвичай швидко прогресують після появи клінічних симптомів.
Всі пріонні захворювання смертельні. Наразі не існує ефективних методів лікування, однак більш глибоке розуміння їх патогенезу в найближчому майбутньому призведе до появи ефективної терапії.
Пріонні захворювання унікальні тим, що можуть передаватися спадково, виникати спорадично або бути інфекційними. Інфекційний агент пріонних захворювань складається з глікопротеїнів аномальної конформації — так званих пріонних білків.
Найперше з цих захворювань було описано скрепі, хворобу овець, яка відома вже більше 250 років. Це захворювання клінічно проявляється підвищеною збудливістю, сильним свербіжем шкіри та атаксією, а в подальшому призводить до паралічу й смерті.
Патофізіологія
Пріонні захворювання клінічно проявляються ураженням нервової системи. Спостерігається вплив на сіру речовину центральної нервової системи, що викликає втрату нейронів, гліоз та характерні губчасті зміни, що представляють собою вакуолізацію нейронів, як показано на зображенні нижче.
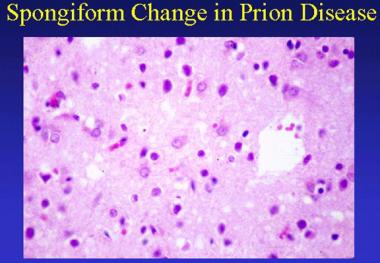
https://emedicine.medscape.com/article/1168941-overview
Крім того, у пацієнтів спостерігаються бляшки з типовими для амілоїду властивостями: наприклад, подвійне променезаломлення яблучно-зеленого кольору після фарбування Конго червоним при перегляді в поляризованому світлі. Приблизно у 10% пацієнтів з ХКЯ амілоїд присутній в мозочку або в півкулях головного мозку.
Етіологія
Щодо складу пріонів було висунуто декілька різних гіпотез. Наукова спільнота підтримала гіпотезу, описану Гриффітом, яку пізніше доповнив Стенлі Прусінер. іпотеза заключалась в тому, що пріони складаються з білку. Прусінер ввів термін «пріон», щоб вказати, що збудником скрепі є білкова інфекційна частинка (PrP).
В 1998 році Прусінер отримав Нобелівську премію за своє відкриття. Його гіпотеза припускає, що пріони не містять нуклеїнової кислоти, а власне патогенний білок було названо PrPSc. Це конформаційно модифікована форма нормального клітинного PrPC, який є білком-господарем, що виявляється на поверхні багатьох клітин, зокрема нейронів. PrPSc при введенні в нормальні здорові клітини викликає перетворення PrPC в PrPSc.
Епідеміологія
Найбільш поширеним з пріонних захворюванням є ХКЯ з частотою приблизно 1 випадок на мільйон населення. Значно рідше зустрічаються сімейні форми пріонних захворювань на зразок ХГШШ та фатального сімейного безсоння. Близько 10% випадків ХКЯ є сімейними, з аутосомно-домінантним типом успадкування, пов'язаним з мутаціями в гені PRNP.
Захворювання, пов'язані з пріонами, невблаганно прогресують і незмінно призводять до смерті.
Середній вік початку спорадичної форми ХКЯ становить 62 роки. Частота захворюваності на спорадичну форму ХКЯ становить близько 1 випадку на мільйон населення, проте серед людей у віці 60-74 років вона становить 5 випадків на мільйон населення. Віковий діапазон може бути широким: випадки були зареєстровані у людей у віці від 17 до 83 років.
Сімейна ХКЯ, ХГШШ і ФСБ мають середній вік початку від 45 до 49 років.
Хвороба Крейтцфельдта-Якоба (ХКЯ)
Спорадична форма ХКЯ (сХКЯ) характеризується мультифокальною неврологічною дисфункцією, що швидко прогресує, міоклонічними судомами, важкими когнітивними порушеннями і смертю приблизно через 8 місяців. Близько 40% пацієнтів з сХКЯ мають когнітивні порушення, 40% — мозочкову дисфункцію, а у решти 20% має місце комбінація цих симптомокомплексів.
Клінічна картина, як правило, також включає поведінкові аномалії, дисфункцію вищої кіркової діяльности, порушення зору, дисфункцію мозочка, а також пірамідні та екстрапірамідні ознаки. Майже у всіх пацієнтів зі сХКЯ розвиваються міоклонічні судоми, що вражають все тіло або кінцівки. Судоми можуть виникати спонтанно чи бути викликані слуховою або тактильною стимуляцією.
У більшості пацієнтів с сХКЯ з'являється характерна картина на ЕЕГ з періодичними або псевдоперіодичними пароксизмами різких хвиль або сплесків на повільному тлі.
Хвороба Герстмана—Штройсслера—Шейнкера (ХГШШ)
ХГШШ була вперше описана в 1936 році. Пацієнти з цим захворюванням страждають на атаксію кінцівок і тулуба, що прогресує, а також на деменцію. Смерть настає через 3-8 років після звернення.
Виражене ураження стовбура мозку часто призводить до симптомів, що вказують на олівопонтоцеребеллярну дегенерацію. Тип успадкування — аутосомно-домінантний. Неврологічні ураження при ХГШШ особливі ще й тим, що відбуваються вони внаслідок значного відкладення амілоїду в ЦНС.
Фатальне сімейне безсоння (ФСБ)
Пацієнти з ФСБ мають важковиліковне безсоння, дизавтономію (гіпертермію, гіпертензію, тахікардію, тахіпное, гіпергідроз), деменцію та моторний параліч. Фенотипічна експресія дуже варіабельна навіть в межах однієї сім'ї.
Вік початку також варіюється від 18 до 60 років. Після появи симптомів курс становить від 6 місяців до 3 років. Через розмаїття клінічних проявів цього розладу генотипування дуже важливо для остаточного діагнозу. Спостерігається виражена атрофія передніх вентральних і медіодорсальних таламічних ядер, що виникає через втрату нейронів та гліоз. На відміну від інших пріонових захворювань, губчаті зміни можуть бути незначними або відсутні зовсім.
Документальный фільм, присвячений пацієнтам з ФСБ
Всі пацієнти з ФСБ мають міссенс-мутацію в кодоні 178 гена PrP, де Asn замінений на Asp в поєднанні з Met в полиморфному кодоні 129.
Причини пріонних хвороб
Пріонні захворювання унікальні тим, що вони можуть бути пов'язані з інфекційними, спорадичними або спадковими причинами.
Інфекційні причини
Куру, одна з форм пріонної хвороби, виникла у племені Форі Східного нагір'я Нової Гвінеї і була пов'язана з ритуальним канібалізмом. Вважається, що захворювання пов'язане з проковтуванням частин тіла пацієнта зі спорадичною формою ХКЯ та з подальшим послідовним розвитком хвороби.
У клінічній практиці ХКЯ передається через хірургічні інструменти, електроди ЕЕГ, трансплантати рогівки, трансплантати твердої мозкової оболонки. Було задокументовано 3 випадки передачі вХКЯ при переливанні крові.
Вважається, що ХКЯ у людей викликається вживанням продуктів з яловичини, контамінованої ГЕВРХ.
Ряд випадків захворювання на спорадичну форму ХКЯ стався в Сполучених Штатах серед молодих людей (<30 років). Захворюваність сХКЯ серед такої вікової категорії історично становила близько 1 випадку на мільярд населення. У 1979-1996 роках в США було зареєстровано 4 випадки сХКЯ серед осіб молодше 30 років. У 1997-2000 роках в США зареєстровано 5 випадків захворювання серед молодих пацієнтів. Двоє з цих осіб прибули з сусідніх округів Мічигану (вік початку захворювання — 26 і 28 років), і 3 випадки сталися серед людей, які були мисливцями на оленів та лосів.
Спадкові причини
Причина виникнення сімейних форм пріонів хвороби пов'язана з мутаціями в гені PrP. Ряд мутацій в гені PrP пов'язаний з аутосомно-домінантними формами хвороби.
Спорадичні випадки
Спорадична ХКЯ — найбільш часта форма хвороби.
Ймовірно, це виникає як спонтанна конформаційна зміна PrPC в форму PrPSc. Форма PrPSc потім саморозповсюджується, спонукаючи більше PrPC перетворювати на PrPSc.
Щоб вивчити зв'язок між медичними процедурами та захворюваністю на спорадичну форму сХКЯ було проаналізовано виконання медичних процедур (хірургічні маніпуляції, нейрохірургічні операції, офтальмологічні операції та переливання крові) у пацієнтів, зареєстрованих Комітетом з нагляду за ХКЯ в Японії з 1999 по 2008 рр. У дослідження були включені 753 пацієнта з сХКЯ і 210 пацієнтів з контрольної групи, а також ті, хто переніс нейрохірургічні або офтальмологічні процедури в самій лікарні. Не було виявлено доказів того, що пріоні хворобі передавалися через досліджувані медичні процедури до початку сХКЯ. Після початку розвитку сХКЯ 4,5% пацієнтів були прооперовані, і ніяких спеціальних запобіжних заходів проти передачі пріонних захворювань не приймалося. Автори прийшли до висновку, що хірургічні процедури або переливання крові мало впливають на частоту сХКЯ.
В даний час встановлена можливість трансфузіонної передачі пріонів. Особи, інфіковані через їжу, можуть передавати інфекційні агенти через здачу донорської крові.
Діагностика
Диференційну діагностику необхідно проводити з:
- негерпесним вірусним енцефалітом,
- дифузною хворобою тілець Леві,
- хронічним менінгітом,
- деменцією як паранеопластичним синдромом,
- сімейною міоклонічною деменцією,
- отруєнням літієм,
- деменцією при захворюваннях рухових нейронів,
- лімбічним енцефалітом (і іншими паранеопластичними синдромами),
- енцефалопатією Хашимото (або стероїд-залежною енцефалопатією, яка пов'язана з аутоімунним тиреоїдитом (SREAT).
Лабораторні дослідження
Щоб виключити токсичну та метаболічну енцефалопатію, необхідно оцінити результати загального аналізу крові, результати функціональних тестів печінки, рівень амонію та швидкість осідання еритроцитів.
При підозрі на бактеріальну інфекцію необхідно виконати посів крові.
Крім того, важливо оцінити функцію щитоподібної залози, рівні B12 та фолієвої кислоти, а також виконати дослідження, щоб виключити нейросифіліс.
Щоб виключити енцефалопатію Хашимото, необхідно визначити сироваткові рівні антитіл до тиреопероксідази.
Якщо можливий паранеопластический синдром (наприклад, коли в анамнезі є злоякісні новоутворення або при дослідженнях виявляють приховані новоутворення), слід дослідити наявність аутоантитіл.
Інструментальні дослідження
МРТ — важливий діагностичний метод. Наприклад, на МРТ можна побачити гіперінтенсивні сигнали в базальних гангліях і таламусі.
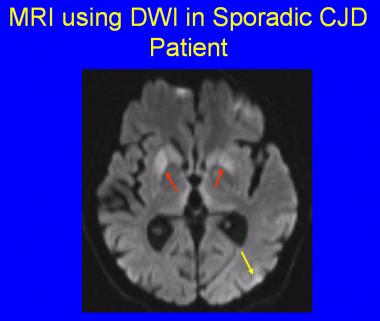
https://emedicine.medscape.com/article/1168941-workup
Описано дві характерні рентгенологічні ознаки. Це ознака «хокейної ключки», яка вказує на посилення сигналу в шкаралупі та голівці хвостатого ядра, що нагадує хокейну ключку, і знак «пульвінар», який відповідає зазвичай двосторонньому посиленому сигналу в ядрах таламусі. Остання ознака є особливо характерною для пацієнтів з вХКЯ.
У деяких пацієнтів була проведена позитронно-емісійна томографія (ПЕТ), при якій був відзначений регіональний гіпометаболізм глюкози, який корелював з невропатологічними ураженнями, виявленими під час розтину.
На додаток до МРТ і ПЕТ, для виявлення змін, характерних для пріоних хвороб, часто слід зробити КТ грудної клітки, черевної порожнини та тазу у пацієнтів, щоб виключити можливість злоякісного новоутворення, яке може викликати паранеопластический синдром.
ЕЕГ
Під час спорадичної ХКЯ у більшості пацієнтів на ЕЕГ з'являються характерні ознаки з періодичними або псевдоперіодичними пароксизмами різких хвиль або спайки на повільному тлі.
При вХКЯ типових змін на ЕЕГ немає.
Лікування
Все пріонні захворювання смертельні. Ефективного лікування наразі немає.
В даний час пацієнтам проводиться симптоматична терапія. При судома призначають протиепілептичні препарати, а пацієнтам з екстрапірамідними симптомами — препарати, які використовуються для лікування хвороби Паркінсона.
В експериментальних системах було показано, що є ряд ліків, ефективний в запобіганні поширенню пріонів. До них відносяться антрацикліни, амфотерицин B і його аналоги, тетрапірроли.
Крім того, дослідження культур тканин показали, що похідні акридину та фенотіазину (наприклад, хінакрін, хлорпромазин) можуть пригнічувати перетворення PrPC в PrPSc. Ці типи ліків використовувалися людьми протягом багатьох років в якості протималярійних та антипсихотичних засобів. Широке клінічне випробування (PRION-1) цього підходу до лікування було розпочато в Великій Британії в 2004 р.
Ще один препарат, що тестується на пацієнтах — полісульфат пентозан. Цого використання в перспективі є багатообіцяючим на підставі досліджень на тваринах. В єдиному описаному клінічному випадку лікування вХКЯ з використанням цього підходу не спостерігалося жодних явних побічних ефектів, і клінічні симптоми, мабуть, були трохи ослаблені, однак за даними КТ атрофія мозку прогресувала.
Інші сучасні підходи включають хелатну терапію. Мідь бере участь у поширенні пріонів, і автори продемонстрували, що хелатор D-пеніциламін затримує початок розвитку пріонної хвороби у інфікованих мишей. In vitro мідь посилювала стійкість пріонів до протеїназ K, чому протидіяла спільна інкубація з D-пеніциліном. В цілому, ці результати показують, що рівень міді може впливати на конформаційний стан PrP, тим самим підвищуючи його інфекційність. Цей ефект можна послабити за допомогою хелатної терапії.
Додатковим терапевтичним підходом, який може бути корисний при пріонних захворюваннях, є імунологічний. У ряді недавніх досліджень було показано, що імунізація пептидами з альфа-спіраллю є дуже успішною в зниженні накопичення церебрального амілоїду.
Профілактика
Так як пріонні захворювання можуть поширюватися ятрогенним шляхом, необхідно бути обережними, щоб не використовувати повторно голки для ЕЕГ та електроміографії, хірургічні та інші інструменти, які використовував пацієнт із пріоною хворобою. Пріонні агенти надзвичайно стійкі до інактивації, а отже, звичайні процедури стерилізації є неефективними.
Прогноз
Громадська організація INgenius – україномовна медична платформа, що пропагує доказову медицину серед спільноти лікарів в Україні з 2016 року. Наша команда створила відкриту базу з перекладених протоколів лікування, аналітичних статей про достовірні методи лікування та розбори фуфломіцинів. Також ми організовуємо на високому рівні події для медиків.
Якщо Ви хочете ще більше доказового україномовного контенту, цікавіших експериментів та практичних заходів, підтримайте нас за допомогою донатів!
Зібрані кошти будуть витрачені на:
- технічне забезпечення сайту;
- щомісячний платіж за платформи такі як ZOOM, telegram і т.д.;
- оплату дизайнера;
- безкоштовні заходи;
- рекламу.
Кожний Ваш внесок - це вклад у майбутнє не тільки наше як платформи, але й також у прогресивний розвиток доказової медицини в України.
Revolution in you!
Кожний Ваш внесок - це вклад у майбутнє не тільки наше як платформи, але й також у прогресивний розвиток доказової медицини в України.
Revolution in you!

info@ingeniusua.org