
Спінальна м’язова атрофія (СМА) — це група спадкових захворювань, які поступово руйнують рухові нейрони, нервові клітини в стовбурі мозку та спинному мозку, які контролюють діяльність скелетних м’язів, таку як розмова, ходьба, дихання та ковтання. Це призводить до м’язової слабкості та атрофії . Рухові нейрони контролюють рух у руках, ногах, грудях, обличчі, горлі та язиці. Коли порушується передача імпульсів між моторними нейронами та м’язами, останні поступово слабшають, починають виснажуватися і розвиваються фасцикуляції (посмикування).
Хвороба вперше описана на початку 1890 -х років Верднігом та Гофманом як прогресуюча м’язова слабкість, що починається у дитячому віці та призводить до ранньої смерті.
Часто діагноз встановлюють надто пізно, що зменшує ефективність лікування та сприяє прогресуванню хвороби. Саме тому, кожному педіатру, сімейному лікарю та батькам, важливо вміти розпізнавати наступні симптоми та вчасно отримувати необхідні консультації спеціалістів:
- слабкі або ослаблені верхні та нижні кінцівки
- рухові проблеми - дітям складно самостійно сідати, повзати чи ходити
- посмикування або тремтіння м'язів (тремор)
- ураження кісток та суглобів - помітні викривлення хребта (сколіоз)
- проблеми з ковтанням, труднощі з диханням
Важливо вчасно помічати "вікове" відставання дитини в фізичному розвитку. Неспроможність дитини самостійно сідати (6-7 місяців), повзяти (9 місяців) стояти з допомогою (9-12 місяців) та ходити (12 місяців та більше), повинні насторожувати батьків та педіатрів. Проте СМА, не спричиняє відставання в інтелектуальному розвитку.
Патофізіологія
Найпоширеніша форма СМА викликана дефектами в обох копіях гена 1 моторного нейрона виживання (SMN1) на хромосомі 5q. Цей ген продукує білок рухових нейронів виживання (SMN), який підтримує здоров'я і нормальну функцію моторних нейронів. Люди зі СМА мають недостатній рівень білка SMN, що призводить до втрати рухових нейронів у спинному мозку, викликаючи слабкість і виснаження скелетних м’язів. Ця слабкість часто є більш вираженою в м’язах тулуба та проксимальних м'язах кінцівок, ніж у кистях і стопах.
Існує багато типів спинномозкової м’язової атрофії, які викликаються змінами одних і тих же генів. Менш поширені форми СМА викликані мутаціями в інших генах, включаючи ген VAPB, розташований у хромосомі 20, ген DYNC1H1 на хромосомі 14, ген BICD2 на хромосомі 9 та ген UBA1 на Х -хромосомі. Типи відрізняються за віком початку та вираженістю м’язової слабкості, однак частково посиндромна картина може мати схожі риси у різних варіантах хвороби.
Як успадковується хвороба?
За винятком рідкісних випадків, спричинених мутаціями гена UBA1, СМА успадковується аутосомно-рецесивним способом, що означає, що уражена людина має два мутовані гени, часто успадковуючи по одному від кожного з батьків. Ті, хто несуть лише один мутований ген, є носіями хвороби без будь -яких симптомів. Аутосомно-рецесивні захворювання можуть вражати кількох людей в одному поколінні (рідні або двоюрідні брати і сестри).

Типи СМА
- СМА типу I, яка також називається хворобою Вердніга-Гофмана, зазвичай виявляється у віці до 6 місяців. Найбільш серйозно хворіють немовлята (СМА типу 0 або IA), які навіть внутрішньоутробно мають зменшені рухи і народжуються з контрактурами та утрудненим диханням, а без лікування смерть настає на першому році життя. Симптоми СМА типу I включають гіпотонію (зниження м’язового тонусу), зменшення рухів кінцівок, відсутність сухожильних рефлексів, фасикуляції, труднощі ковтання, а також порушення дихання. У цих дітей з віком також розвивається сколіоз або інші аномалії скелета. Без лікування хворі діти ніколи не сидять і не стоять, і переважна більшість зазвичай помирає від дихальної недостатності у віці до 2 років.
- У дітей із формою СМА II типу перші симптоми зазвичай проявляються у віці від 6 до 18 місяців, хоча деякі можуть проявлятися раніше. Вони можуть сидіти без опори, але не можуть стояти або ходити без сторонньої допомоги, а деякі можуть втратити здатність сидіти самостійно з часом без лікування. Такі хворі можуть мати проблеми з диханням, включаючи гіповентиляцію уві сні. Захворювання невпинно прогресує без лікування. Тривалість життя таких дітей скорочена, але більшість доживає до підліткового або молодого віку.
- У дітей із СМА III типу (хвороба Кугельберга-Веландера) симптоми з’являються після 18-місячного віку і такі діти можуть самостійно ходити. Вони спочатку виявляють труднощі при ходьбі і бігу, підйомі по сходах або вставанні зі стільця. Найчастіше спочатку уражаються проксимальні м’язи ніг, в руках спостерігається тремтіння. Ускладнення включають сколіоз та суглобові контрактури — хронічне скорочення м’язів або сухожиль навколо суглобів, викликане аномальним м’язовим тонусом і слабкістю, що перешкоджає суглобам вільно рухатися. Люди зі СМА III типу можуть бути схильні до респіраторних інфекцій, але більшість із них має нормальну тривалість життя.
- У осіб із СМА IV типу симптоми розвиваються після 21 року, характеризуються легкою чи помірною проксимальною м’язовою слабкістю.
Діагностика
Необхідним є аналіз крові для виявлення делецій або мутацій гена SMN1. Цей тест ідентифікує принаймні 95 відсотків СМА типів I, II та III, а також може виявити, чи є людина носієм дефектного гена, який можна передати дітям. Якщо не виявлено аномалій гену SMN1 або анамнез та обстеження особи не є типовими для SMA, інші діагностичні тести можуть включати електроміографію (реєстрація електричної активності м’язів під час скорочення та в спокої), дослідження швидкості нервової провідності (визначення здатності нерва посилати електричний сигнал), біопсію м’язів (використовується для діагностики багатьох нервово-м’язових розладів) тощо.
Лікування
Не можливо повністю вилікувати СМА. Лікування полягає в усуненні симптомів і запобіганні ускладнень.
У грудні 2016 року Управління з контролю за продуктами та ліками США затвердило нузінерсен як перший препарат, схвалений для лікування дітей та дорослих зі СМА. Препарат вводять шляхом ін’єкції в рідину, що оточує спинний мозок. Механізм його дії полягає у збільшенні вироблення повнорозмірного білка SMN, що має вирішальне значення для підтримки рухових нейронів.
Найкращі результати задокументовані у немовлят та дітей, в яких якомога раніше розпочато лікування.
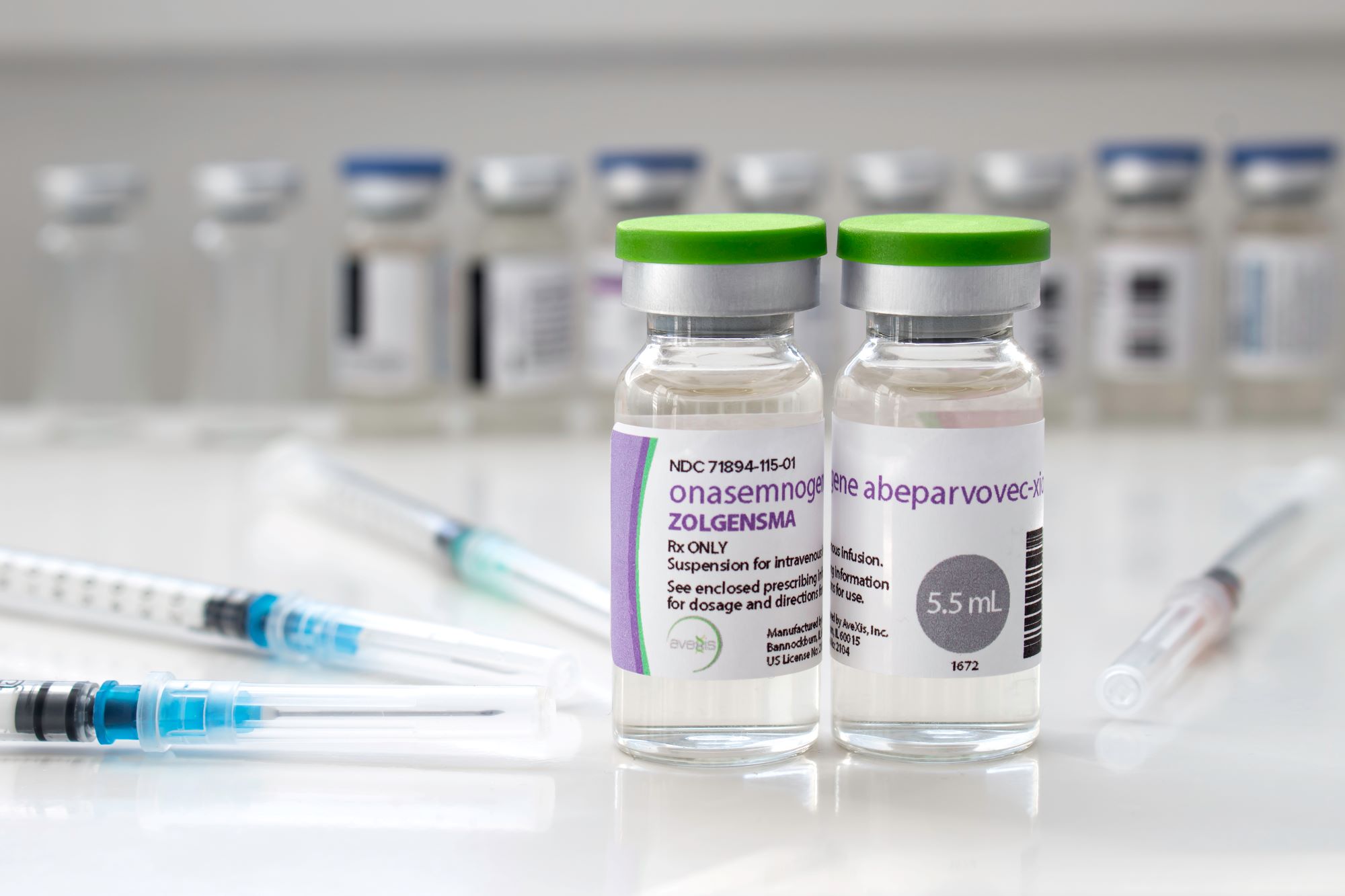
У травні 2019 року FDA схвалила генну терапію препаратом Zolgensma для дітей віком до 2 років, які мають СМА І типу. Безпечний вірус доставляє повністю функціональний ген SMN людини до цільових мотонейронів, що, у свою чергу, покращує рух і функцію м’язів, а також покращує виживання.
Вартість препарату Zolgensma становить близько 2 мільйонів доларів. У 2019 компанія-розробник заявила, що буде розігрувати приблизно 100 доз на рік для дітей у країнах, де продаж препарату поки що не дозволено.
У серпні 2020 року FDA схвалила пероральний препарат рисдиплам (Evrysdi™) для лікування пацієнтів у віці від двох місяців і старше із СМА.
Додаткова терапія
Фізична реабілітація може допомогти поліпшити поставу, запобігти контрактурі суглобів, уповільнити м’язову слабкість та атрофію. Вправи на розтяжку можуть допомогти збільшити діапазон рухів і підтримувати циркуляцію крові. Іноді потрібна додаткова терапія для подолання труднощів з мовленням та ковтанням. Допоміжні пристрої, такі як брекети, ортопедичне взуття, синтезатори мовлення та сучасні обладнані візки можуть допомогти забезпечити життя без сторонньої допомоги.
Прогноз
Прогноз змінюється залежно від типу СМА. Деякі форми СМА є смертельними без лікування.
Люди зі СМА можуть здаватися стабільними протягом тривалих періодів, але поліпшення без лікування не слід очікувати.
СМА в Україні
В Україні створено сайт, на якому зібрали та уніфікували інформацію про дітей, хворих на спінальну м'язову атрофію (СМА) - https://vstygnemo.org.ua
На сайті є детальна інформація про кожну дитину зі СМА в Україні і є можливість зробити пожертву.
The non-governmental organization INgenius is a Ukrainian-language medical platform that has been promoting evidence-based medicine among the community of doctors in Ukraine since 2016. Our team has created an open database of translated treatment protocols, analytical articles on reliable treatments, and analysis of fuflomycins. We also organize events for doctors on a highly professional level.
If you want even more evidence-based Ukrainian-language content, more interesting experiments, and practical activities, support us with the help of donors!
The funds raised will be spent on:
- technical support of the site;
- the monthly payment for platforms such as ZOOM, telegram, etc.;
- payment for a designer;
- free events;
- advertising.
Each of your contributions is a contribution to the future not only of our platform but also of the progressive development of evidence-based medicine in Ukraine.
Revolution in you!

ingeniusua@gmail.com

